This page was generated from
docs/Examples/CrystallisationTests/BasalticCrystallisation.ipynb.
Interactive online version:
![]() .
.
Modelling liquid-line-of-descents
One of the primary uses of the MELTS models is simulating the crystallisation of magma under different conditions.
Useful for identifying whether samples are linked by crystallisation or if other processes are required to explain the geochemical variability of a region.
Before any calculations can be run users need to download the alphaMELTS for MATLAB files (https://magmasource.caltech.edu/gitlist/MELTS_Matlab.git/) and store them locally on their computer. These files then need to be added to the Python path by using the \(sys.path.append()\) command below.
Data used in the calculations below can be downloaded from here: https://github.com/gleesonm1/pyMELTScalc/blob/master/docs/Examples/CrystallisationTests/Fernandina_glass.xlsx
[1]:
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import pyMELTScalc as M
import sys
sys.path.append(r"MELTS")
In this notebook we’ll show how pyMELTScalc can quickly perform multiple calculations (utilizing parallel processing), and investigate how the liquid-line-of-descent is linked to the conditions of magma storage.
First, we need to load in some data. For this example we’ll use data from Isla Fernandina in the Galapagos Archipelago, where a series of melt inclusions (Koleszar et al. 2009) and matrix glasses (Peterson et al. 2017) provide us with plenty of glass data to constain the magmatic evolution of the sub-volcanic system. The melt inclusion and matrix glass data is included in a single excel spreadsheet that we can load in using Pandas. We can also split this DataFrame into two. One containing just the melt inclusion data, the second including just the matrix glass data.
[2]:
df = pd.read_excel('Fernandina_glass.xlsx')
df = df.fillna(0)
# split data based on the Group (Melt Inclusion or Matrix Glass)
MI = df.loc[df['Group'] == 'MI',:].reset_index(drop = True)
MG = df.loc[df['Group'] == 'MG',:].reset_index(drop = True)
MG.head()
[2]:
| Group | Latitude | Longitude | SiO2 | TiO2 | Al2O3 | FeOt | MnO | MgO | CaO | Na2O | K2O | P2O5 | H2O | CO2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | MG | -0.17 | -91.77 | 49.11 | 2.90 | 14.04 | 11.38 | 0.21 | 6.53 | 11.88 | 2.55 | 0.43 | 0.30 | 0.593 | 0.011442 |
| 1 | MG | -0.17 | -91.77 | 49.22 | 3.10 | 14.09 | 11.37 | 0.20 | 6.75 | 11.78 | 2.75 | 0.44 | 0.31 | 0.683 | 0.009890 |
| 2 | MG | -0.21 | -91.81 | 49.01 | 2.92 | 13.92 | 11.74 | 0.21 | 6.31 | 11.90 | 2.55 | 0.46 | 0.33 | 0.635 | 0.012210 |
| 3 | MG | -0.21 | -91.80 | 49.42 | 3.14 | 13.92 | 11.77 | 0.20 | 6.47 | 11.46 | 2.82 | 0.46 | 0.33 | 0.732 | 0.010531 |
| 4 | MG | -0.24 | -91.75 | 48.76 | 3.57 | 13.88 | 11.83 | 0.23 | 6.37 | 11.40 | 3.09 | 0.52 | 0.36 | 0.834 | 0.007797 |
One of the key things that we need to perform any calculations with MELTS is a starting composition. To start, let’s use the most primitive (highest MgO) composition from the melt inclusion analyses. There are a few ways to find this composition, the way I like to do so (not necessarily the quickest) is to simply sort the melt inclusion DataFrame by the MgO contents and select the first row:
[3]:
MI = MI.sort_values('MgO', ascending = False, ignore_index = True)
MI.head()
[3]:
| Group | Latitude | Longitude | SiO2 | TiO2 | Al2O3 | FeOt | MnO | MgO | CaO | Na2O | K2O | P2O5 | H2O | CO2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | MI | 0.0 | 0.0 | 47.5 | 2.29 | 16.4 | 9.16 | 0.123 | 9.38 | 11.6 | 2.25 | 0.329 | 0.257 | 0.68 | 0.0280 |
| 1 | MI | 0.0 | 0.0 | 47.5 | 2.58 | 15.5 | 10.04 | 0.138 | 8.97 | 11.4 | 2.44 | 0.428 | 0.262 | 0.69 | 0.0630 |
| 2 | MI | 0.0 | 0.0 | 49.2 | 2.33 | 16.0 | 8.67 | 0.147 | 8.94 | 12.0 | 1.78 | 0.005 | 0.094 | 0.78 | 0.0004 |
| 3 | MI | 0.0 | 0.0 | 48.4 | 2.50 | 15.7 | 9.07 | 0.139 | 8.88 | 11.6 | 2.18 | 0.398 | 0.326 | 0.69 | 0.0126 |
| 4 | MI | 0.0 | 0.0 | 48.1 | 2.46 | 15.3 | 8.96 | 0.153 | 8.82 | 12.1 | 2.34 | 0.325 | 0.387 | 0.88 | 0.4986 |
[4]:
starting_comp = MI.loc[0]
starting_comp
[4]:
Group MI
Latitude 0.0
Longitude 0.0
SiO2 47.5
TiO2 2.29
Al2O3 16.4
FeOt 9.16
MnO 0.123
MgO 9.38
CaO 11.6
Na2O 2.25
K2O 0.329
P2O5 0.257
H2O 0.68
CO2 0.028
Name: 0, dtype: object
We can now perform crystallisation calculations. These calculations are run using rhyoliteMELTSv1.2.0 (we have some CO\(_2\) in the starting composition), start at the liquidus and end at 1100\(^o\)C. The calculations are also run at 1 log unit below the FMQ buffer. By specifying Frac_solid = True, we inform pyMELTScalc that we want to perform a fractional (rather than equilibrium) crystallisation scenario.
[5]:
Isobaric_Xtal = M.isobaric_crystallisation(Model = "MELTSv1.2.0",
bulk = starting_comp,
find_liquidus = True,
P_bar = np.array([500,1000,2000,4000]),
T_end_C = 1100,
dt_C = 2,
fO2_buffer = "FMQ",
fO2_offset = -1.0,
Frac_solid = True,
Frac_fluid = True,
label = "pressure")
Running MELTSv1.2.0 calculations 0 to 3.0 ... Complete (time taken = 86.84 seconds)
dict_keys(['P = 1000 bars', 'P = 2000 bars', 'P = 4000 bars', 'P = 500 bars'])
This code will perform all 4 calculations at the specified pressures simultaneously. This ‘parallel processing’ significantly reduces the time required to perform these models.
There are several ways to display the results. Option 1 is to use the in-built harker plotting function which directly plots a 3-by-2 group of plots without having to make any changes to the output from the crystallisation models. You’ll see below that the code also allows other data to be loaded and plotted alongside the models:
[6]:
M.harker(Results = Isobaric_Xtal, data = df, label = True)
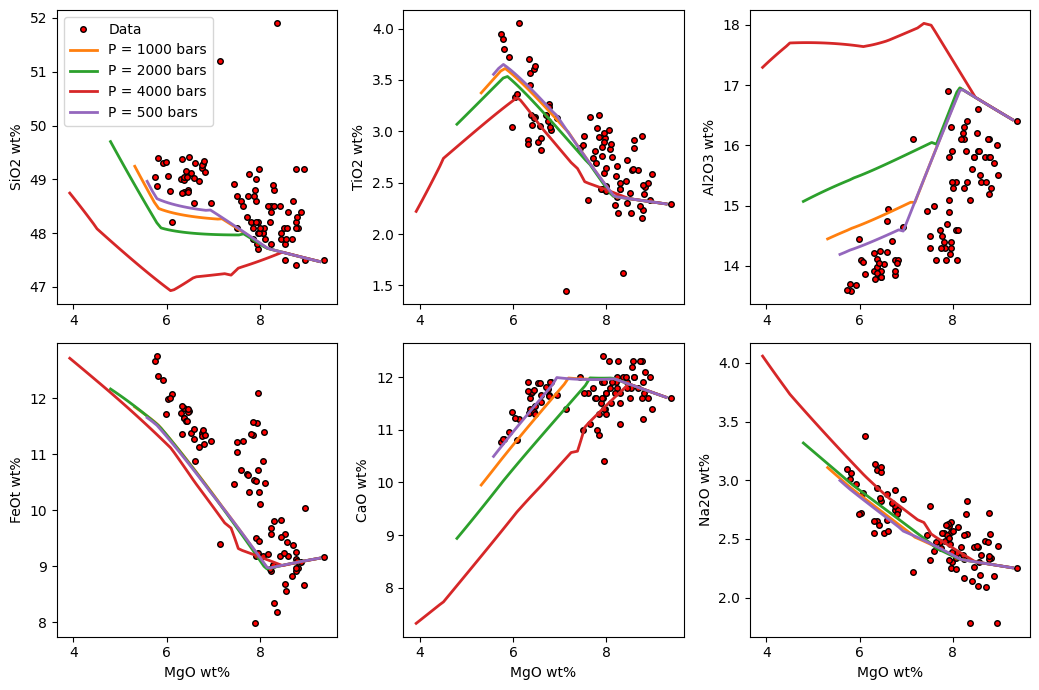
The problem, is that this code doesn’t distinguish between the melt inclusion and matrix glass data. If we want to do that it is relatively straightforward to create a small snippet of code to plot the compositional variation of these magmas:
[7]:
x_axis = 'MgO'
y_axis = ['Al2O3', 'FeOt', 'CaO']
f, a = plt.subplots(1,3, figsize = (12,4), sharex = True)
for i in range(len(y_axis)):
a[i].plot(MI[x_axis], MI[y_axis[i]], 'ok', mfc = 'b', label = "Melt Inclusions")
a[i].plot(MG[x_axis], MG[y_axis[i]], 'sk', mfc = 'y', label = "Matrix Glass")
for r in Isobaric_Xtal:
a[i].plot(Isobaric_Xtal[r]['All'][x_axis + '_Liq'],
Isobaric_Xtal[r]['All'][y_axis[i] + '_Liq'],
'--', label = r)
a[i].set_xlabel(x_axis)
a[i].set_ylabel(y_axis[i])
a[2].legend()
[7]:
<matplotlib.legend.Legend at 0x293f1171070>
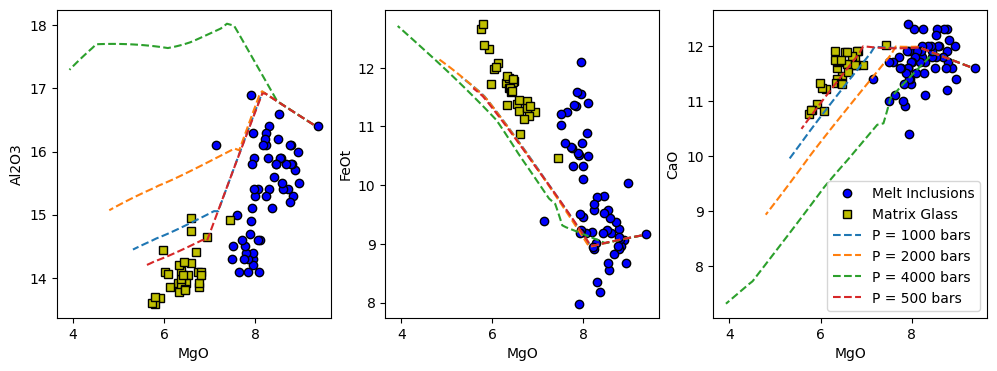
We can see that the lower pressure models generally provide a better fit to the Data. Also, examining the results would reveal that as pressure decreases, clinopyroxene stability decreases (it saturates at lower MgO contents) as would be expected based on experimental data.
There are, however, several other variables that we haven’t explored in these models above that might influence our results. To demonstrate this, let’s change the melt H\(_2\)O and CO\(_2\) contents and re-run the models.
Gleeson et al. (2022) suggest that the melt inclusion H\(_2\)O contents measured by Koleszar et al. (2009) overpredict the melt H\(_2\)O content at the time of melt inclusion formation. In addition, the melt CO\(_2\) content was likely underestimated as the CO\(_2\) content in melt inclusions from Fernandinas reaches up to around 5000 ppm. Therefore, let’s use these values to see how changing melt H\(_2\)O and CO\(_2\) contents influences the liquid line of descent. There is no need to change our starting composition, we can simply use the H2O_Liq and CO2_Liq inputs in the isobaric_crystallisation function to ‘overwrite’ the initial H\(_2\)O and CO\(_2\) contents of the magma.
[8]:
Isobaric_Xtal_CO2 = M.isobaric_crystallisation(Model = "MELTSv1.2.0",
bulk = starting_comp,
find_liquidus = True,
P_bar = np.array([500,1000,2000,4000]),
T_end_C = 1100,
dt_C = 2,
fO2_buffer = "FMQ",
fO2_offset = -1.0,
Frac_solid = True,
Frac_fluid = True,
H2O_Liq = 0.4,
CO2_Liq = 0.5,
label = "pressure")
Running MELTSv1.2.0 calculations 0 to 3.0 ... Complete (time taken = 141.63 seconds)
dict_keys(['P = 1000 bars', 'P = 2000 bars', 'P = 4000 bars', 'P = 500 bars'])
[9]:
x_axis = 'MgO'
y_axis = ['Al2O3', 'FeOt', 'CaO']
f, a = plt.subplots(1,3, figsize = (12,4), sharex = True)
for i in range(len(y_axis)):
a[i].plot(MI[x_axis], MI[y_axis[i]], 'ok', mfc = 'b', label = "Melt Inclusions")
a[i].plot(MG[x_axis], MG[y_axis[i]], 'sk', mfc = 'y', label = "Matrix Glass")
for r in Isobaric_Xtal_CO2:
a[i].plot(Isobaric_Xtal_CO2[r]['All'][x_axis + '_Liq'],
Isobaric_Xtal_CO2[r]['All'][y_axis[i] + '_Liq'],
'--', label = r)
a[i].set_xlabel(x_axis)
a[i].set_ylabel(y_axis[i])
a[2].legend()
[9]:
<matplotlib.legend.Legend at 0x293eec15880>
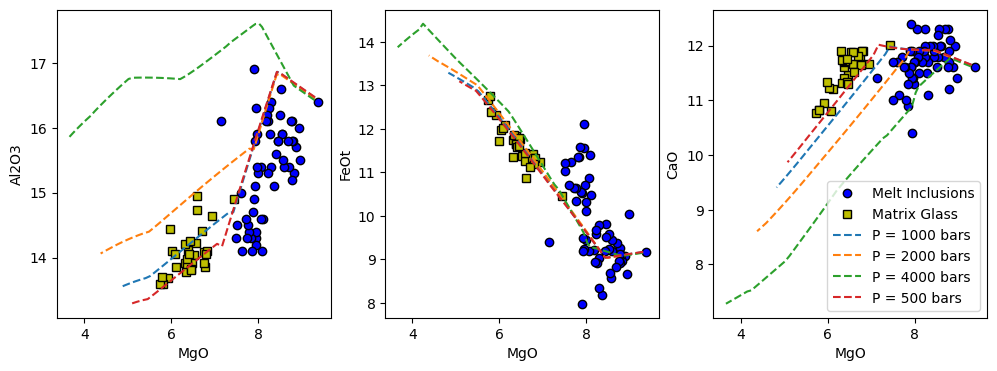
We can see that making this change (at least for Al2O3 and FeOt) has improved the fit between the model predictions and the matrix glass data. This demonstrates that there are multiple variables that influence the crystallisation path predicted by MELTS.